CORE CAPABILITY
Alignments + QC
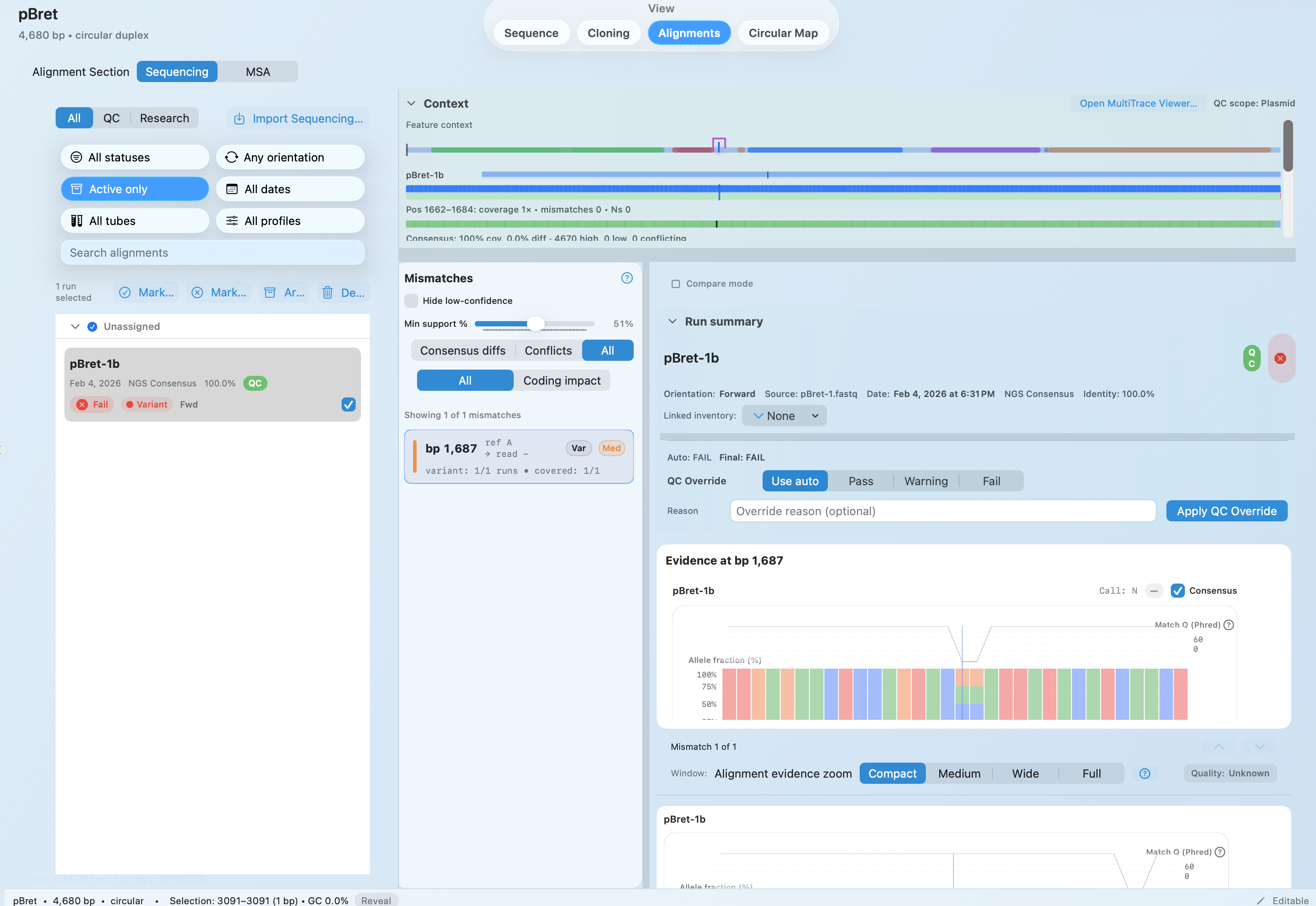
MolBioCove aligns Sanger traces and FASTQ/consensus reads back to the expected construct sequence, calls mismatches automatically, and pins evidence (trace, quality, allele fraction) to the exact bases—so you can confirm true variants, ignore noise, and keep QC decisions reviewable. Local-first, natively built on macOS.
PRODUCT VIEW
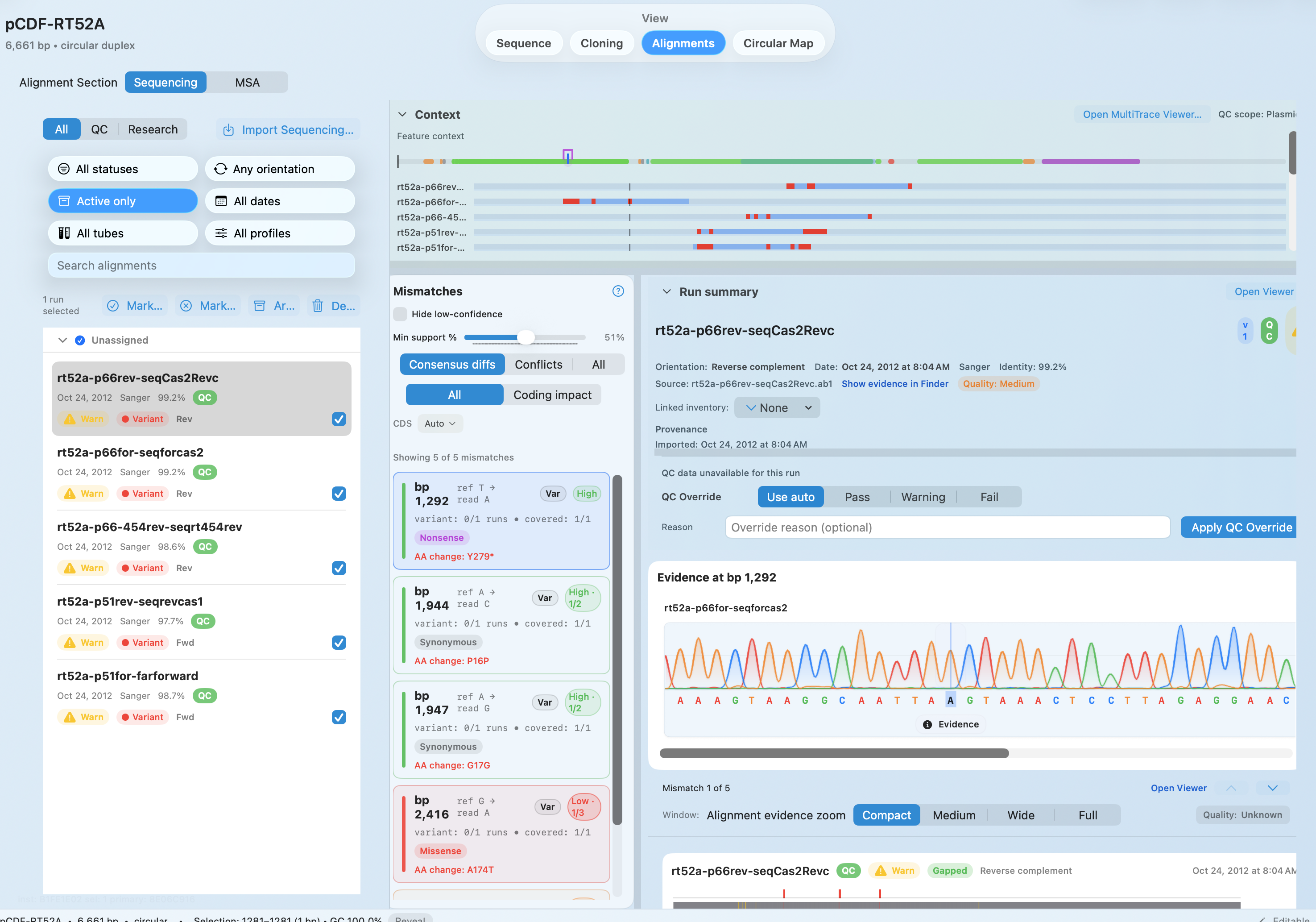
Sanger mismatch detection with coding impact
Automatic calls highlight what changed—and whether it affects the coding sequence—so you can focus on the mismatches that matter.
EVIDENCE‑LINKED REVIEW
Go from mismatch → evidence in one click
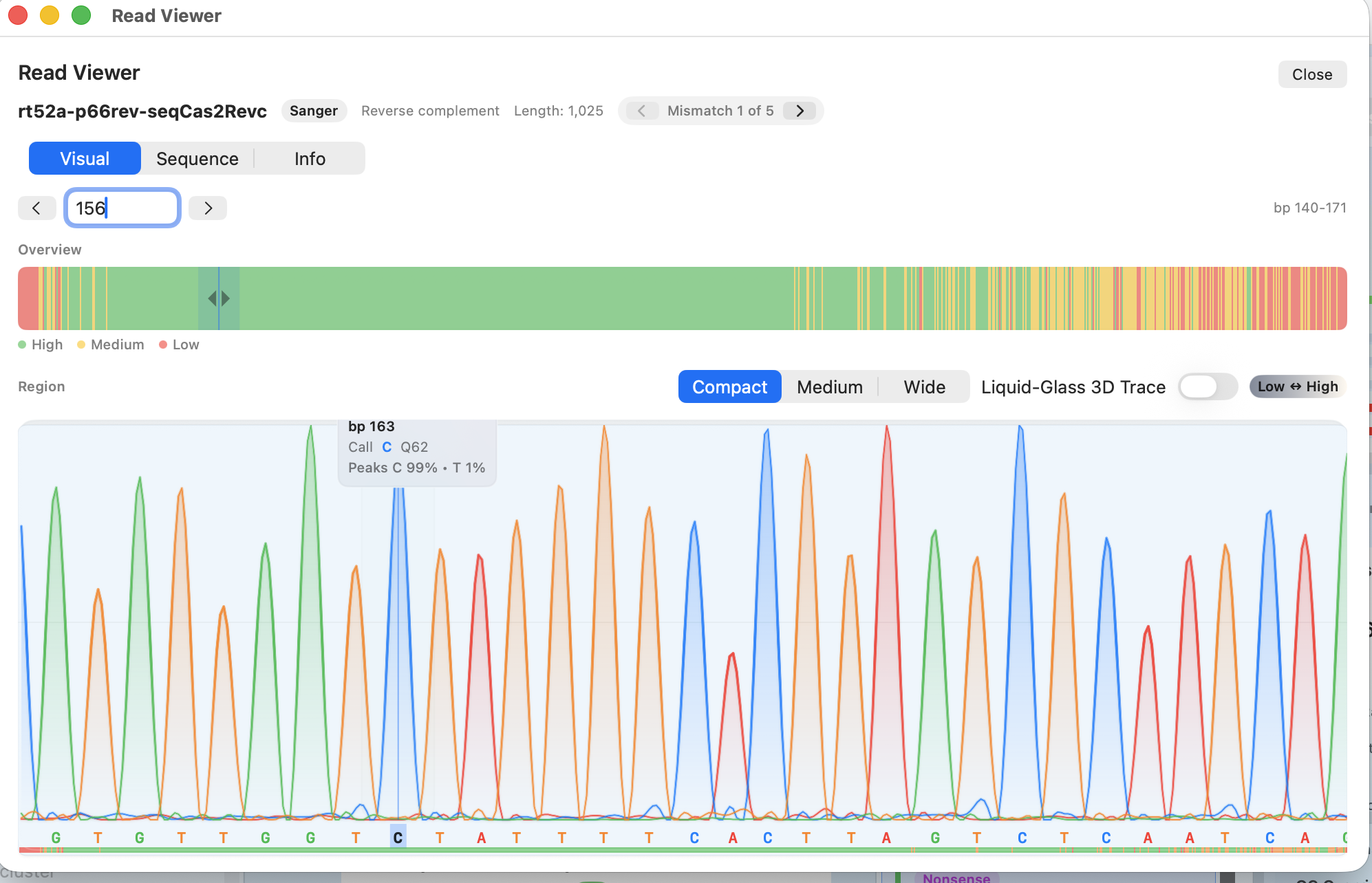
Individual read viewer
Step through mismatches, inspect chromatograms, and confirm the signal before you act.
- Next/previous mismatch navigation with filters
- Trace, base calls, and quality alongside the reference
- Copy a clean 5′→3′ sequence or capture notes for the run
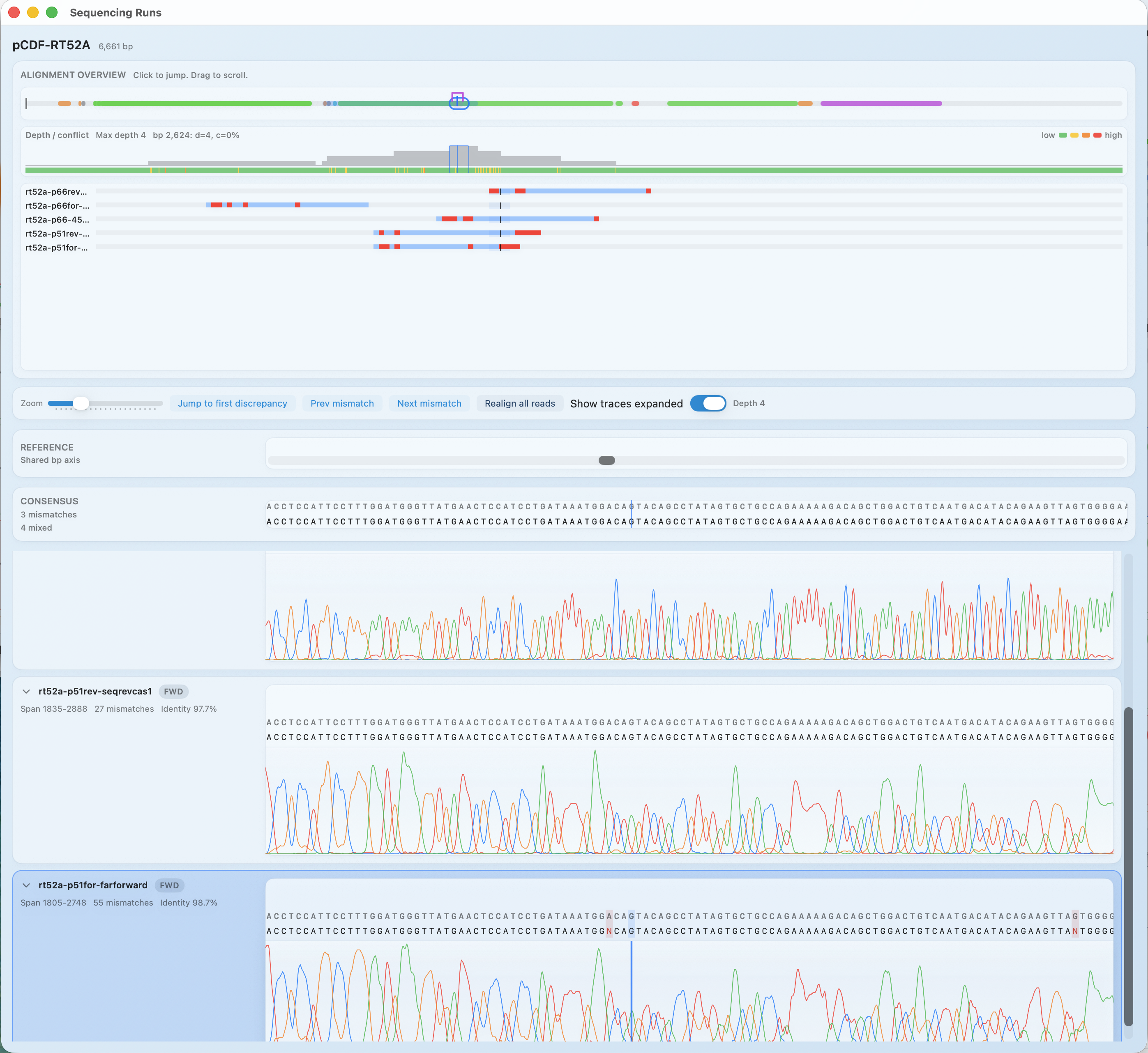
Sequencing runs overview
Compare multiple reads against the same construct and see where they agree, conflict, or drop in quality.
- Conflict and coverage overview across the full sequence
- Jump to the first mismatch across runs
- QC summaries per read/run, stored with the construct
FASTQ + CONSENSUS
Validate NGS reads with base‑level evidence
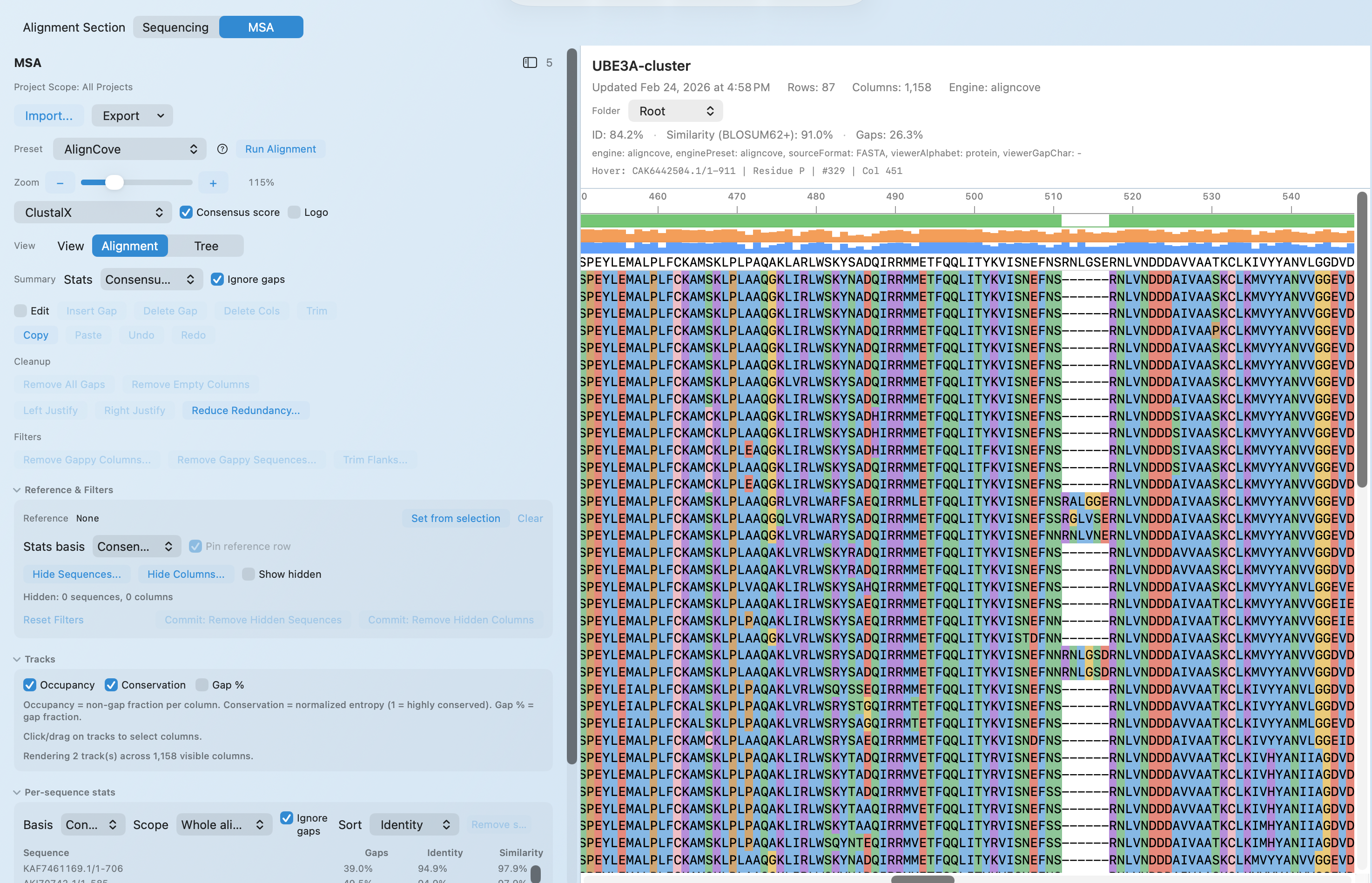
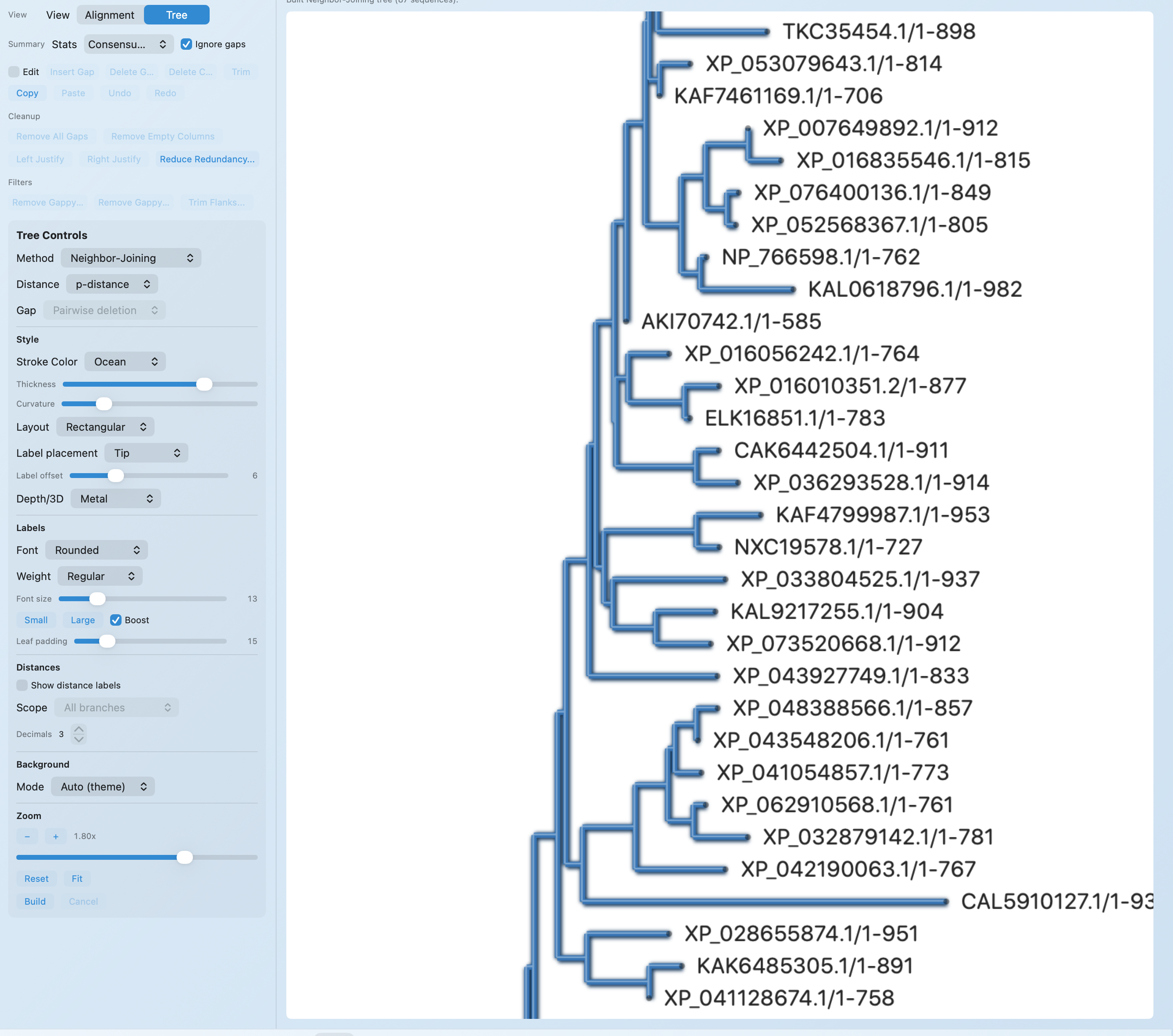
MULTIPLE SEQUENCE ALIGNMENT
AlignCove: deterministic MSA for remote‑homology proteins
AlignCove is MolBioCove’s default protein multiple‑sequence alignment (MSA) engine, tuned for challenging remote‑homology families. It’s deterministic by design (same input → same output), so results are reproducible and easy to compare across runs and collaborators. No ML inference—fully algorithmic and deterministic.
How it works (high level)
- Input normalization + QA
- Difficulty stats (sequence count, lengths, similarity)
- Guide tree from k‑mer distances (k=6)
- Progressive alignment (sequence↔sequence, sequence↔profile, profile↔profile; affine‑gap DP)
- Pairwise rescue candidates (only for the final two‑sequence merge)
- Stage‑3 refinement (bounded local search; deterministic tie‑breaks)
- Output + diagnostics (final alignment + run metadata)
AlignCoveV1 is the stable shipping preset. Today, aligncove and aligncove‑v1 produce identical results for the same input (and remote7 is a historical alias).
When AlignCoveV1 collapses to a final two‑sequence merge, it can evaluate a small, deterministic set of “rescue” candidates (e.g. diagonal‑biased DP and terminal‑gap rebalance). A rescue is used only if it improves the unbiased score and passes safety checks (to avoid pathological alignments).
Alignment pipeline schematic

From input normalization to refinement, with optional pairwise rescue evaluated only when the pipeline reaches a final two‑sequence merge.
Benchmarks (alignment quality; quality‑only)
On a curated set of 13 difficult OXBench families (SP‑F1; higher is better):
- AlignCoveV1 mean SP‑F1: 0.6337
- Best external tool per family (mean SP‑F1): 0.7053 (gap: 0.0715)
External tool mean SP‑F1 (same set):
- T‑Coffee: 0.6758
- ProbCons: 0.6620
- MUSCLE: 0.6587
- MAFFT: 0.6572
- Clustal Omega: 0.6511
- MSAProbs: 0.5822
Note: this comparison is quality‑only; it does not include external tool runtimes.
WHAT YOU CAN DO
Common tasks
TYPICAL WORKFLOW
A typical flow
Add Sanger or FASTQ runs and attach them to the construct you’re validating.
Align against the expected construct sequence with mismatch calls kept in-context.
Use evidence-linked views to confirm true variants and ignore noise.
Export a QC record (and keep history) so handoffs stay reviewable.
OUTPUTS
Outputs
Request Beta Access
Join the external beta—built for active bench teams.