AB1 file viewer plus chromatogram evidence
Open AB1 files against the expected construct and jump to the bases that need a decision.
AB1 VIEWER + VALIDATION
Open AB1 files, review chromatograms, check FASTQ-backed calls, and rerun AlignCove protein MSA with the same output every time.
OUTCOMES
Open AB1 files against the expected construct and jump to the bases that need a decision.
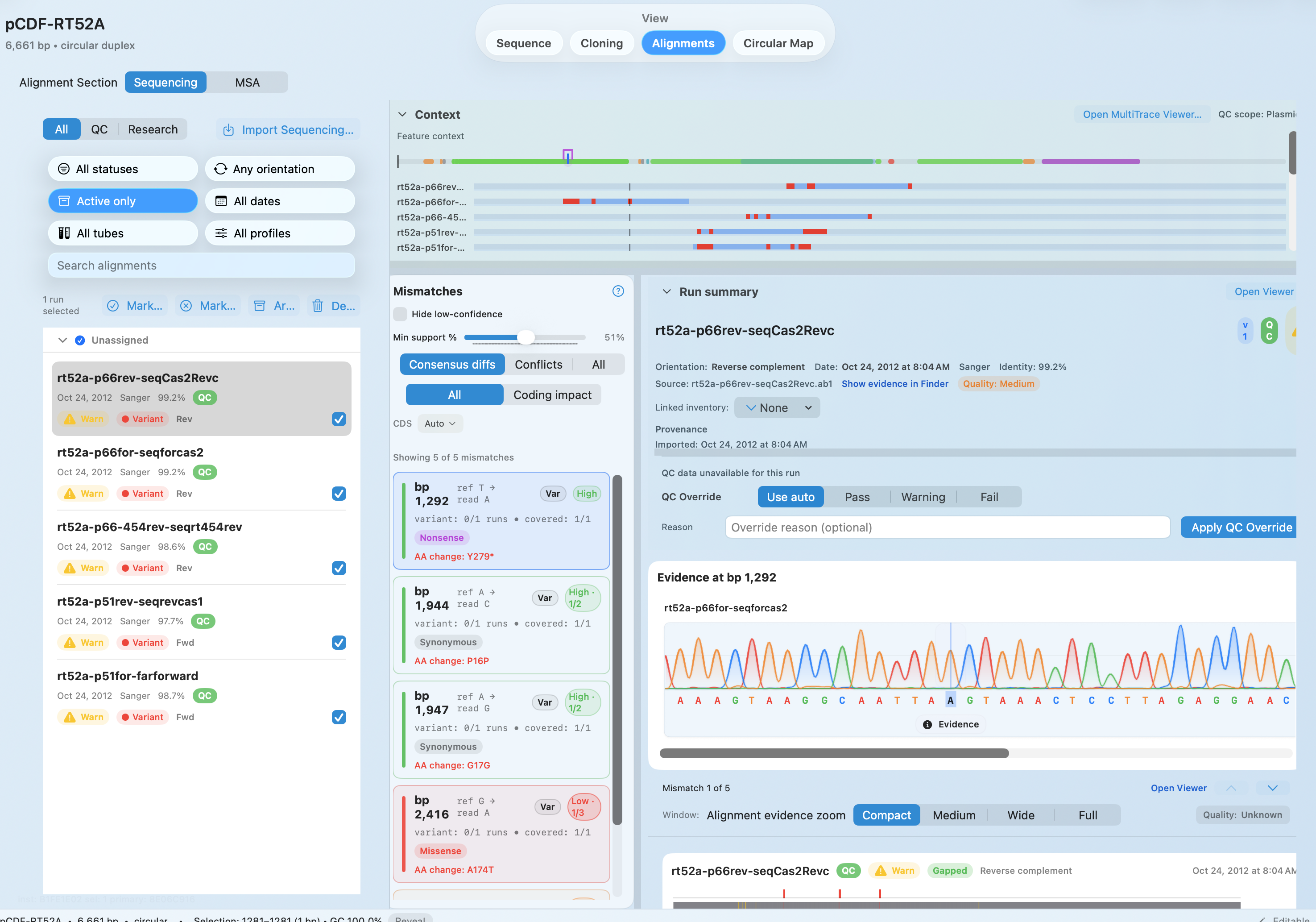
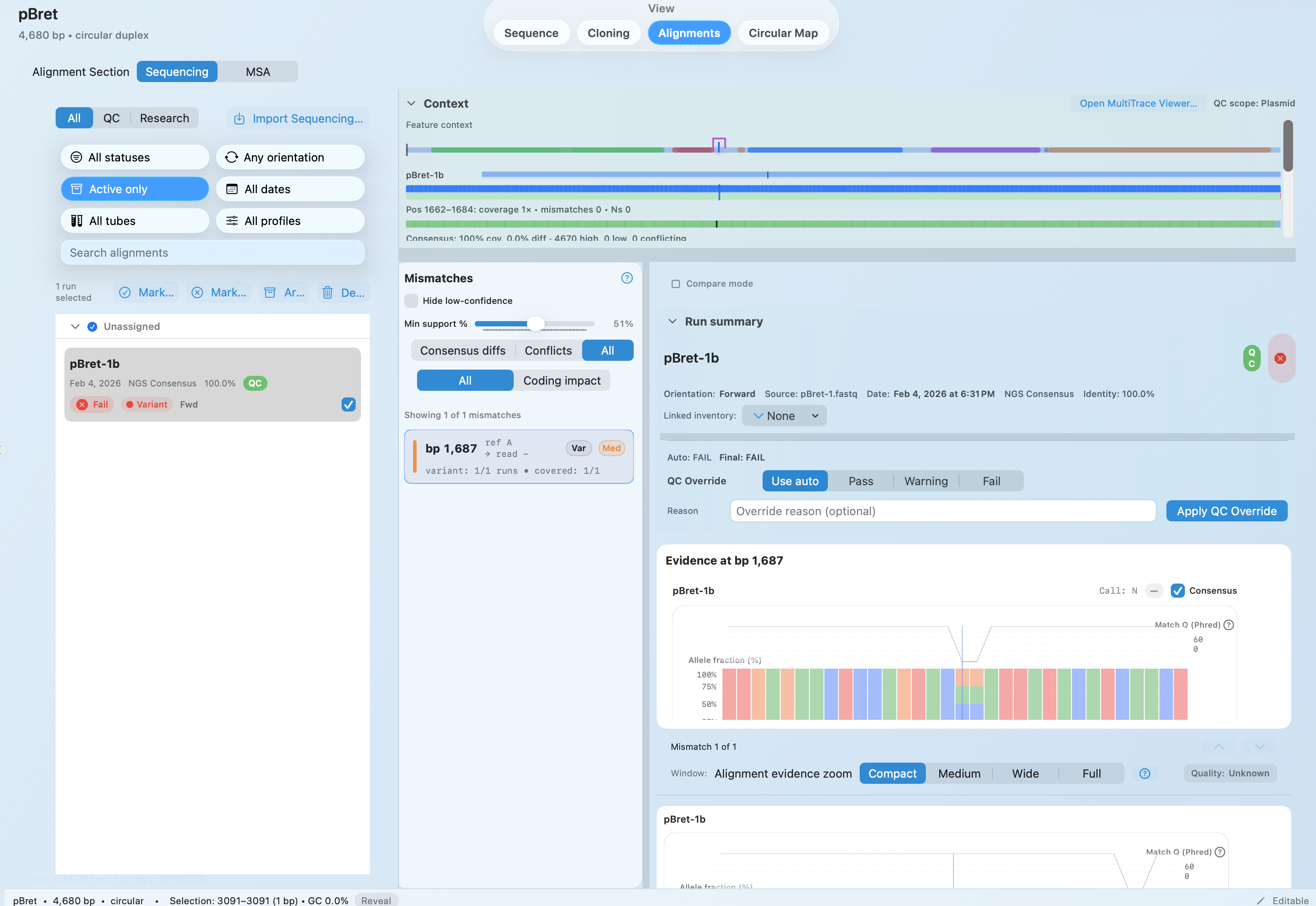
See how much support each FASTQ mismatch has before you decide whether it is real.
Keep validation outcomes, comments, and follow-up history with the same construct record.
Repeatable MSA matters when you compare variants across experiments or need a stable methods story.
SEQUENCING VALIDATION
Use RayCrest as Sanger sequence analysis software with construct-aware mismatch review and coding-impact context from the start.
.ab1 and .abi.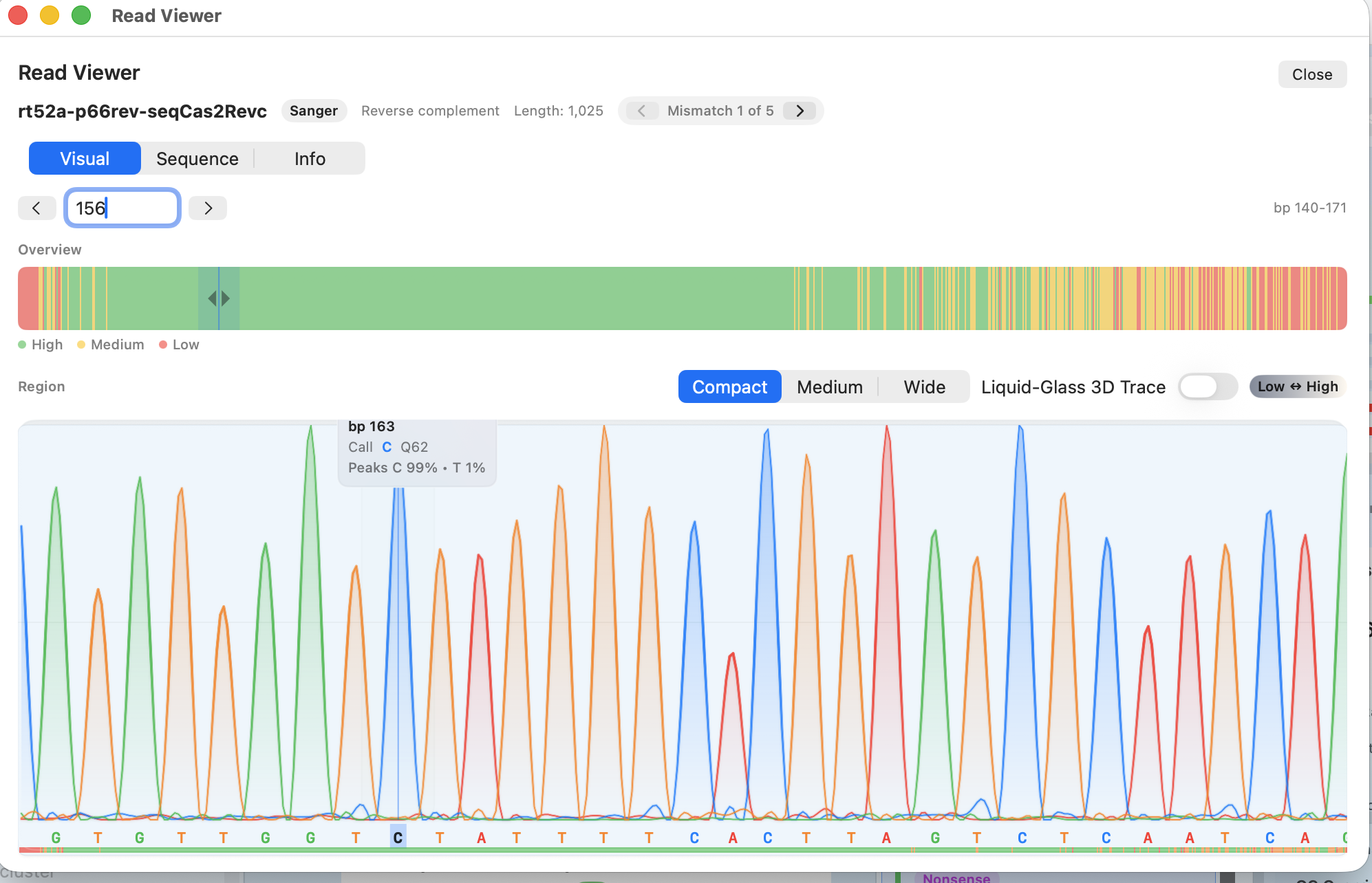
Move from a flagged mismatch to review chromatograms and supporting evidence without switching tools.
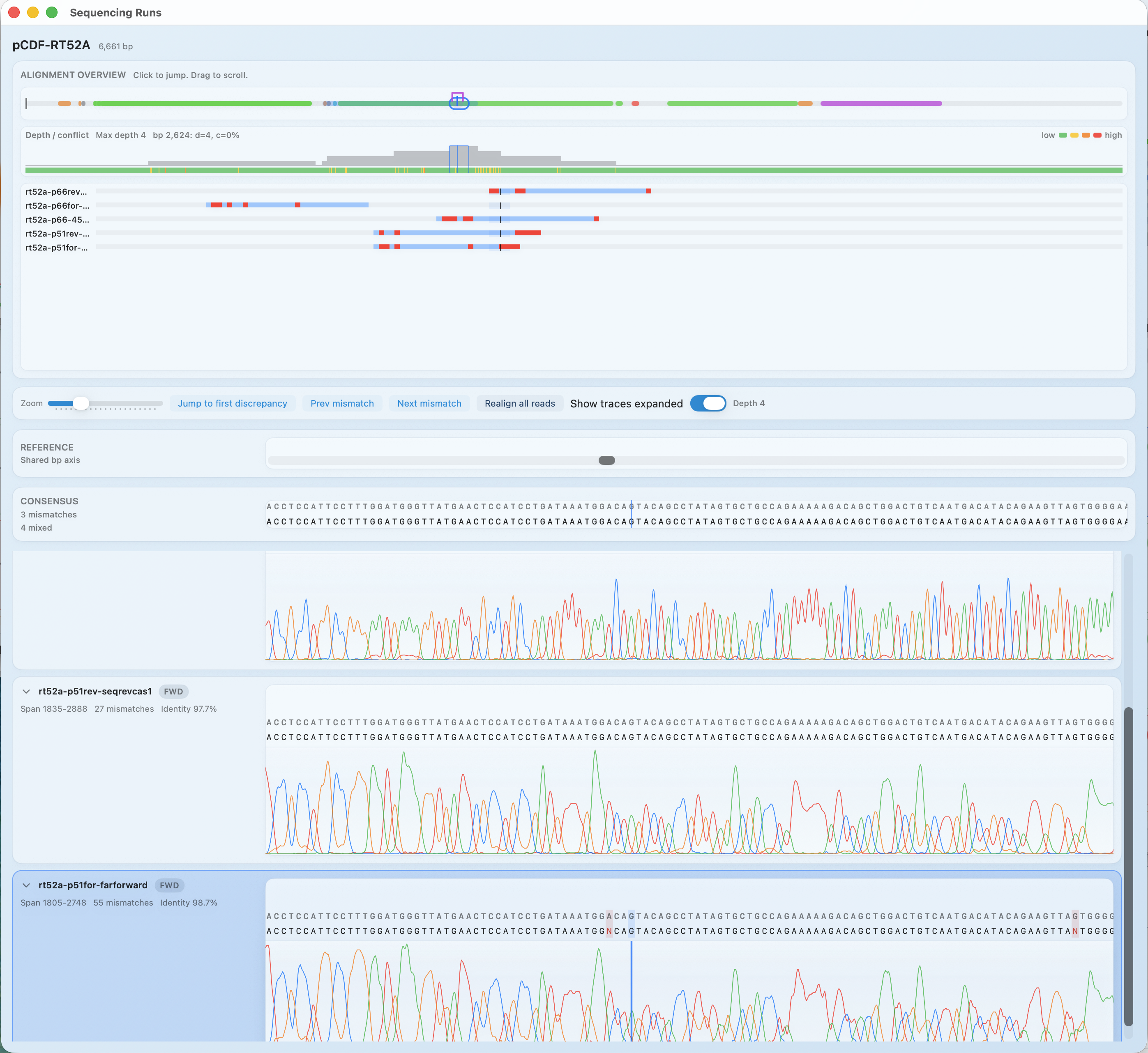
Compare multiple runs against one construct, resolve disagreements, and keep the decision history where teammates can find it.
When a base looks off, the built-in FASTQ viewer keeps read support and QC context at the exact position under review.
MULTIPLE SEQUENCE ALIGNMENT
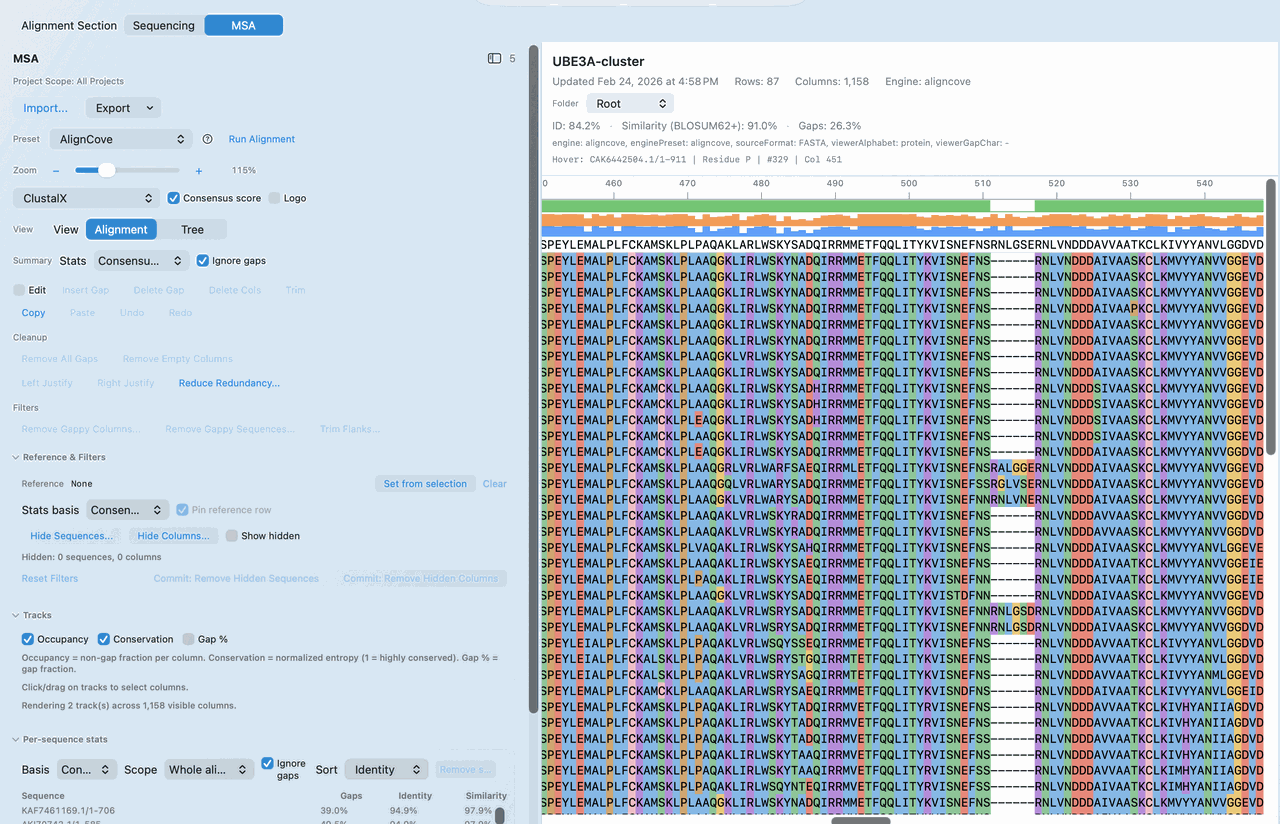
Most teams don't need just any alignment result. They need one they can rerun later and still compare directly against earlier decisions, figures, and methods.
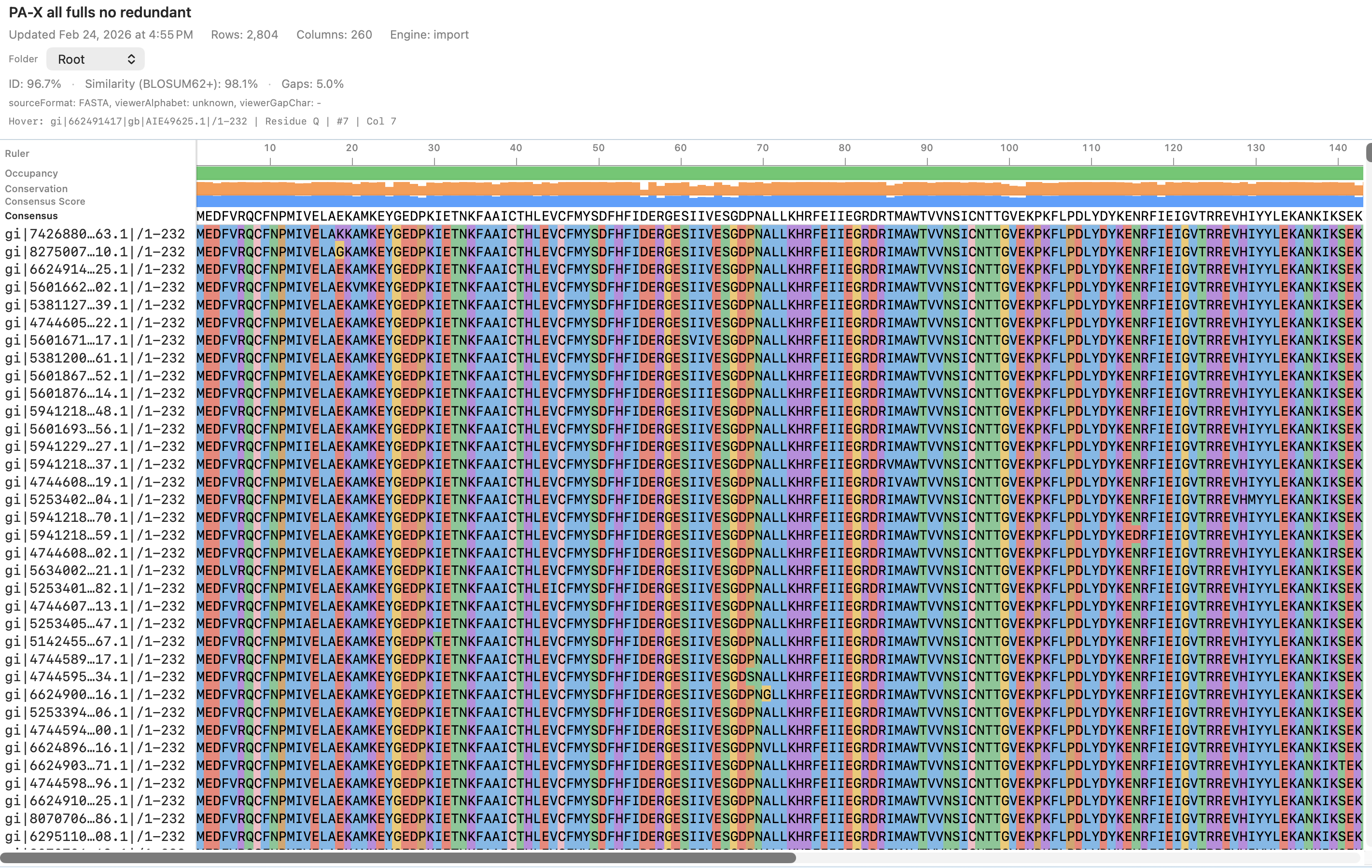
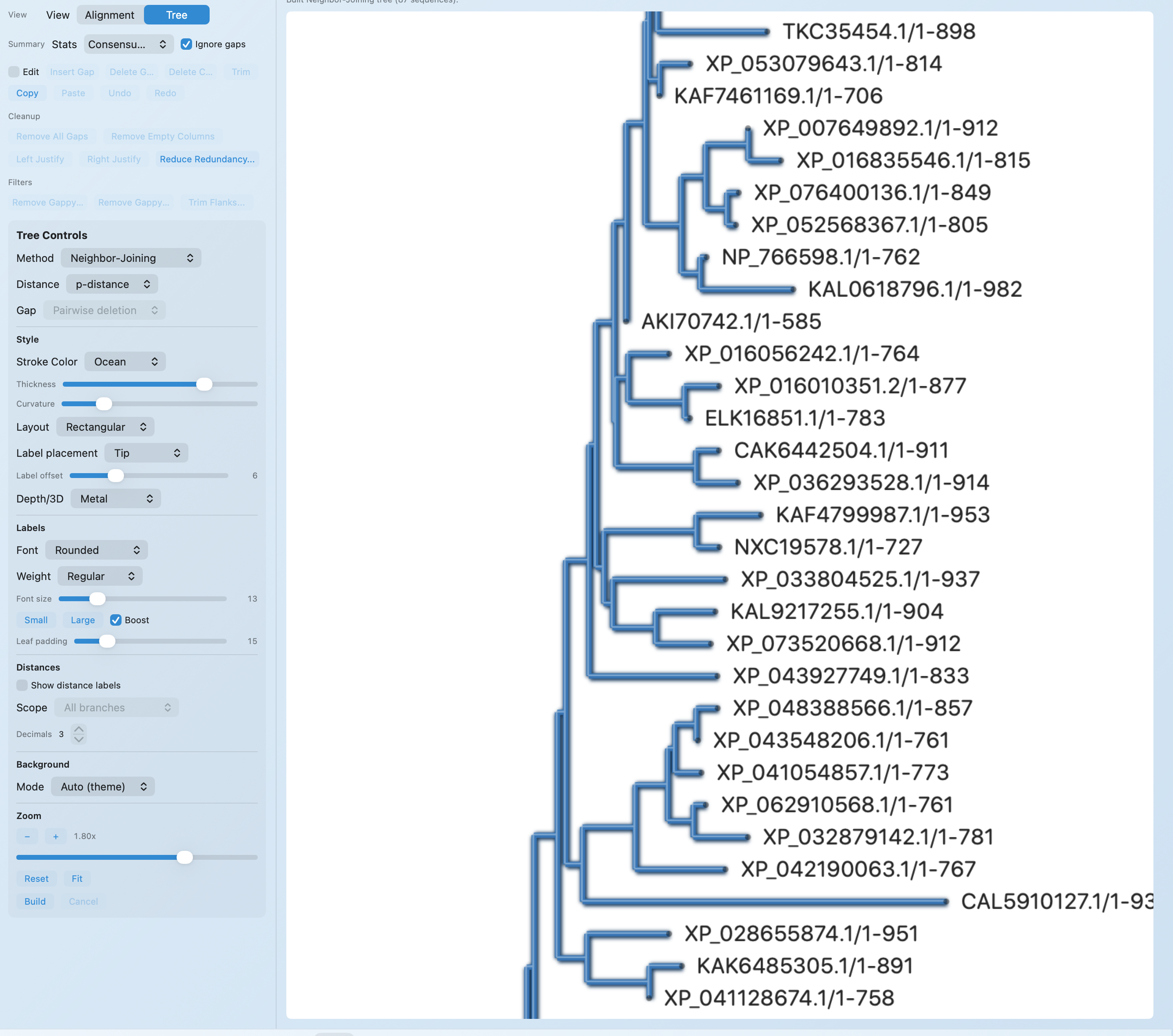
AlignCove is RayCrest’s built-in protein alignment tool, so you can inspect the alignment itself, switch to the tree view, and keep run diagnostics nearby.
TECHNICAL DETAILS
The validation story is centered on evidence-linked review, not just on a final pass or fail label.
.ab1 and .abi..fastq, .fq, and .fastq.gz.AlignCove uses a multi-stage process tuned for difficult protein families, especially the distantly related ones where standard tools often produce shaky alignments.
The preset behavior is locked down so repeated runs stay comparable, and the rescue step is constrained rather than open-ended.
AlignCoveV1 is the stable shipping preset. Today, aligncove and aligncove-v1 produce identical results for the same input (with remote7 retained as a historical alias).
When the workflow reaches a final two-sequence merge, a small deterministic rescue set may be evaluated (for example, diagonal-biased DP and terminal-gap rebalance). Rescue is accepted only if the unbiased score improves and safety checks pass.
No ML inference is used in this pipeline; behavior is algorithmic and deterministic.
AlignCove performs comparably to established tools like T-Coffee and MAFFT on difficult protein families, landing within about 7% of the best-per-family score while also giving reproducible output on repeat runs.
On a curated set of 13 difficult OXBench families (SP-F1, higher is better):
External tool mean SP-F1 on the same set: T-Coffee 0.6758, ProbCons 0.6620, MUSCLE 0.6587, MAFFT 0.6572, Clustal Omega 0.6511, MSAProbs 0.5822.
This comparison is quality-only and does not include external runtime measurements.
Once the run finishes, you can inspect the alignment itself or switch to a tree view without exporting the data into another tool first.
WORKFLOW + OUTPUTS
Add Sanger .ab1/.abi traces or FASTQ runs and attach them to the expected construct.
Navigate mismatch calls, inspect chromatogram or FASTQ support, and confirm evidence at read/base resolution.
Mark the construct as passed, flagged, or failed so the decision and the evidence behind it stay saved together.
RELATED NEXT STEPS
AB1 Viewer
Use the focused landing page when you need the AB1 file viewer, chromatogram viewer, and construct-linked validation story in one place.
MSA
Get the plain-English AlignCove story, plus viewer, tree view, and determinism messaging tuned for protein MSA searches.
Access
Tell us if AB1 review, FASTQ-backed decisions, or repeatable protein MSA is the workflow your team needs first.
Construct Editing
Move from evidence review back into the construct workspace for edits, primer redesign, and the next cloning pass.
RayCrest MDS is available now for macOS 13+.